In Silico studies
Mutation frequency and protein structure
To assess the likelihood of pathogenicity, missense mutations are first screened against large databases such as the EXAC, SFARI and the ClinVar and LOVD database as well as in-house databases,
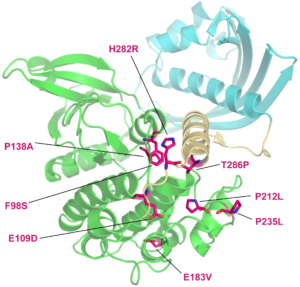
To investigate how missense variants affect protein structure, we use prediction programs such as PolyPhen and SuSPect and structural protein data. To obtain structures for the proteins (or protein domains) of interest, we use the mapping file pdbtosp.txt from UniProt. If PDB structures are not available, structural models will be generated using Phyre2 and I-TASSER protein structure prediction servers. Models will be generated for the reference (wild-type) and mutant sequences. High confidence models will be used for structurally alignment with PyMol to assess the predicted effect on the conformation of the protein.